Revolutionary development in the field of battery technology through innovative sulphur cathodes
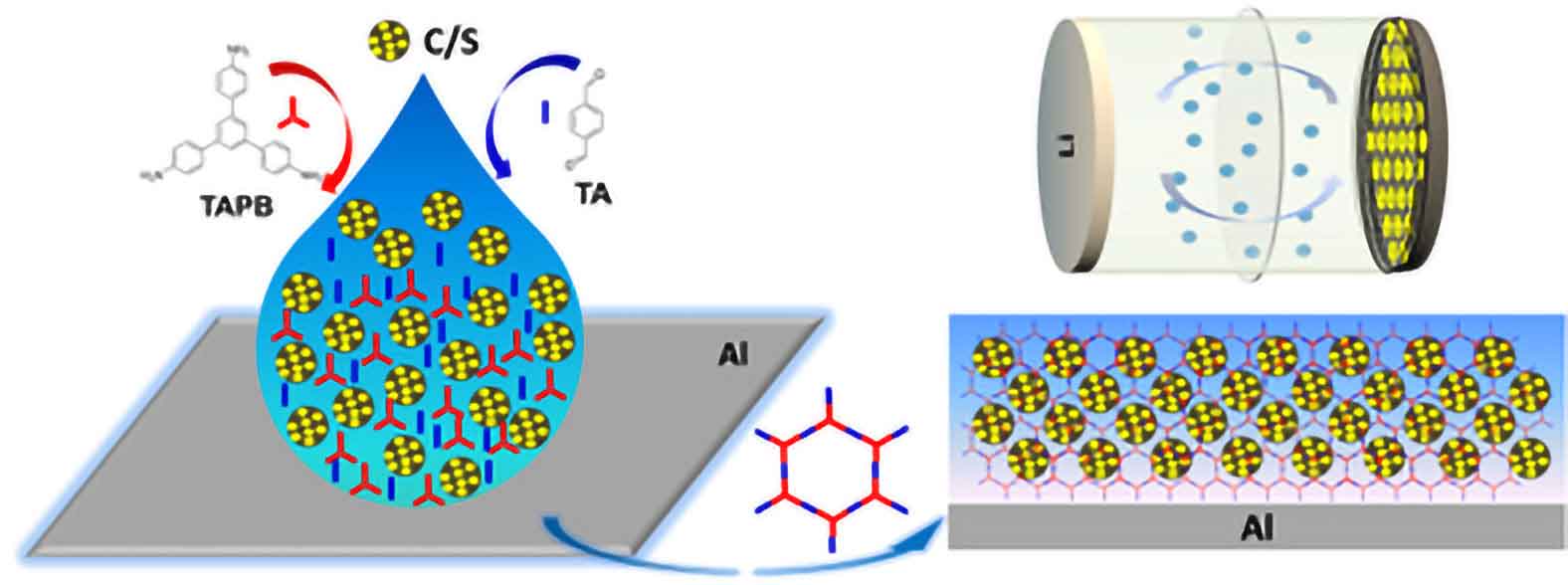
Innovative research has brought us closer to sustainable battery technology with a breakthrough in sulfur-based cathodes. Traditionally, lithium-ion batteries — central to electronics and electric vehicles — rely on scarce materials like cobalt. Sulfur offers a greener alternative due to its abundance and impressive theoretical capacity of 1675 mAh g-1.
A major challenge with sulfur has been the “sulfur-shuttle” effect, where sulfur’s mobility leads to rapid battery degradation. However, a recent study introduces a novel solution: encapsulating sulfur within a microporous, imine-based polymer network directly on the current collector. This one-pot synthesis approach not only streamlines production but also signicantly boosts battery performance.
This innovative cathode design enables selective electrolyte and Li-ion transport while robustly containing the sulfur, delivering high performance across discharge rates — from 1360 mAh g-1 at 0.1 C to 807 mAh g-1 at 3 C. Advanced analysis through DFT calculations and operando Raman spectroscopy has shown that the polymer’s imine groups enhance polysulde binding, effectively reducing degradation.
This breakthrough paves the way for sulfur-based cathodes to become a viable alternative to metalbased ones, marking a signicant step toward greener, high-performance battery technologies. Keep an eye on this space — sulfur could be the future of batteries!
One-pot Synthesis of High-capacity Sulfur Cathodes via In-situ Polymerization of a Porous Imine-based Polymer
Guiping Li, Ye Liu, Thorsten Schultz, Moritz Exner, Ruslan Muydinov, Hui Wang, Kerstin Scheurell, Jieyang Huang, Norbert Koch, Paulina Szymoniak, Nicola Pinna, Philipp Adelhlem, Michael Janus Bojdys
Angew. Chem. Int. Ed., e202400382 (2024) OPEN ACCESS ACCESS
DOI: 10.1002/anie.202400382
Electrically controlled straining of a suspended 2D semiconductor drives different energy states into hybridization, enabling single photon emission

Semiconducting, two-dimensional transition metal dichalcogenides (TMDs) have attracted much interest during the last 15 years. Their highly tuneable mechanical and optoelectronic properties and their combinability with other 2D materials into new structures make TMDs promising systems for many technologies such as electronics and power generation and storage. Their rich low dimensional physics include exotic phenomena as Bose-Einstein condensation or single photon emission, some of them not yet fully explained. Light-matter interaction in 2D semiconductors is governed by excitons, Coulomb-bound electron-hole pairs that can be classified as bright or dark depending on how strongly they couple with light. Despite being the ground state of the system, dark excitons in WSe2 are now only starting to be systematically studied. The group "Physics of Low Dimensional Systems" of Dr. Sebastian Heeg in HU Berlin in collaboration with AG Bolotin at FU Berlin and AG Libisch at TU Vienna have investigated the strain-dependence of dark excitons in monolayer WSe2. This system is particularly relevant as many studies have reported single photon emitters in strained WSe2 but failed to explain the microscopic mechanism underneath quantum light emission.
Dr. Heeg explains how this project was originally conceived: “Our colleagues in the group of Florian Libisch from TU Vienna published in 2019 a very suggesting paper where they predicted that strain in single layer WSe2 could bring dark excitons in resonance with naturally occurring defect energy levels. Once the two excitonic species were tuned into the same energy, they would combine their properties creating a hybridized state that enables single photon emission. When we read about this theoretical model, we immediately decided we wanted to probe it experimentally. To do so, we developed an electrostatic straining technique that allowed us to apply strain controllably at low and high temperatures on WSe2 membranes while probing their electronic structure with photoluminescence spectroscopy.”
By applying strain on WSe2 membranes with a high degree of control, the authors characterized the different excitonic species present in the material according to their energetic dependence with strain. At certain strain levels, strain-independent defect states and strain dependent dark excitons became energy degenerated and their photoluminescence intensity increased an order of magnitude. More evidence for the hybridization of the state such as avoided-crossing energy shifts and temperature tunability of the strain necessary to trigger hybridization was also reported. “The high tunability of the hybridized state enabled by our device architecture will be likely key for the operation of single quantum emitters in WSe2” highlights Pablo Hernández López, a PhD candidate at Dr. Heeg’s group and co-author of the paper, “On the other hand, the characterization and tuning of the energy hierarchy of the excitons present on suspended materials using our electrostatic straining approach opens the door for more exciting discoveries in the future”.
Strain control of hybridization between dark and localized excitons in a 2D semiconductor
P. Hernández López, S. Heeg, C. Schattauer, S. Kovalchuk, A. Kumar, D.J. Bock, J.N. Kirchhof, B. Hoefer, K. Greben, D. Yagodkin, L. Linhart, F. Libisch, K.I. Bolotin
Nature Communications, 13, 7691 (2022) OPENACCESS
DOI: 10.1038/s41467-022-35352-9
New production method for flexible, durable anodes with high capacity-to-weight
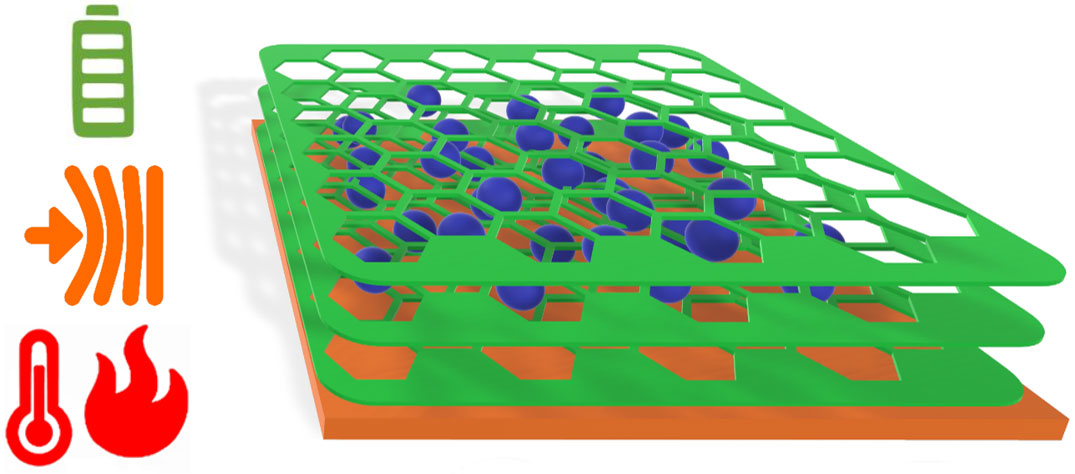
A team of researchers from Humboldt Universität zu Berlin, the Leibniz-Institut für Polymerforschung Dresden (IPF) e. V., and the Karlsruhe Institute of Technology (KIT) have made an anode with superior performance for portable battery applications, close to limits of theoretical capacity. Uniquely, the obtained anodes are flexible without surface reconstruction or crack formation, and they survive heat shocks without performance-loss.
Conventional batteries break down when subjected to mechanical and thermal stress. As a necessity, they need to be situated in stiff, rigid sections of nominally “foldable” electronics and away from sources of heat. The fundamental limitation of the conventional production methods is that the free motion of binders and additives used for battery assembly will lead, over time, to a detrimental loss of the desired electrochemical bias, and finally to a dead battery. To overcome this limitation, the team of Prof. Michael J. Bojdys, the team leader at Humboldt Universität zu Berlin, had the idea to replace conventional binders and additives with a semi-conducting porous organic polymer that (i) adheres to the current collector and grows around the active material, and (ii) enables transport of electrolyte and charge-carriers.
Prof. Bojdys says “Batteries work because we carefully build a chemical bias from small particles. You see this represented in the plus-pole and minus-pole on the battery. Now, what happens if you start shaking or heating such an ordered system? Well, you destroy the chemical bias, and the battery is dead! The way how we build conventional batteries is comparable to putting all your shopping loosely into your car trunk – by the time you get home, everything is jumbled up. Now, if you want to keep your shopping ordered, you obviously put it into bags! This is the role that our semi-conducting porous polymer plays in our electrodes. The polymer replaces all classic battery additives, and it leads to fantastic performance.”
Based on this technology Dr. Goshtasp Cheraghian and Prof. Dr. Michael J. Bojdys seek to commercialize their electrodes and ink-formulations as part of the INAM AdMaLab 2022 Incubator Program.
The article appeared online as:
One‐pot synthesis of high‐capacity silicon anodes via on‐copper growth of a semiconducting, porous polymer
J. Huang, A. Martin, A. Urbanski, R. Kulkarni, P. Amsalem, M. Exner, G. Li, J. Müller, D. Burmeister, N. Koch, T. Brezesinski, N. Pinna, P. Uhlmann, and M.J. Bojdys
Natural Sciences, published online (2022) OPENACCESS
DOI: 10.1002/ntls.20210105
FAIR research data in materials sciences
The FAIRmat consortium led by Humboldt-Universität zu Berlin describes its concept for accessible research data in the renowned journal Nature
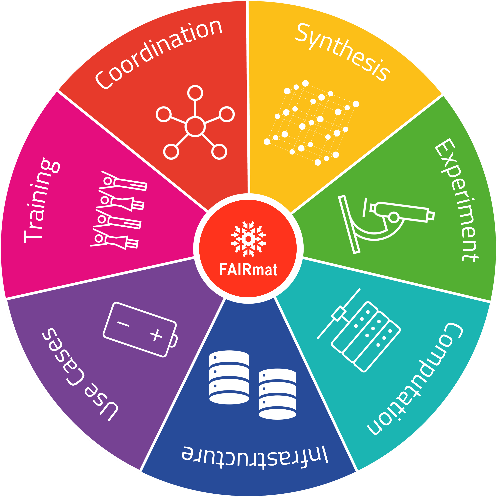
How the FAIRmat consortium works (Copyright: FAIRmat)
The lifestyle of our society is largely determined by the achievements of condensed matter physics, chemistry and material sciences. Touch screens, batteries, electronics or implants: Many new products in the fields of energy, environment, health, mobility and information technology are largely based on improved or even novel materials. These fields generate enormous amounts of data every day, which is a new raw material - and is therefore a gold mine in itself. However, a prerequisite for this is that these data are comprehensively characterized and made available to science.
FAIR data for shared use
The FAIRmat consortium ("FAIR Data Infrastructure for Condensed-Matter Physics and the Chemical Physics of Solids"), led by IRIS Adlershof member Prof. Claudia Draxl, aims to refine this raw material, i.e. turning data into knowledge and value. A prerequisite is a data infrastructure that makes enables scientific data to be „FAIR", i.e. findable, accessible, interoperable and re-purposable. "With a "FAIR" infrastructure, data can be easily shared and explored using data analytics and artificial intelligence methods. This approach will significantly change the way how science is done today," says Claudia Draxl.
In the journal Nature, the scientists describe how such a data infrastructure can be successfully implemented in the field of materials science. The article appears today in the "Perspectives" format, in which the journal publishes forward-looking contributions that stimulate discussion and new scientific approaches.
The FAIRmat consortium is part of the initiative “Nationale Forschungsdaten Infrastruktur” (NFDI). The project is based on the extensive experience with the world's largest data infrastructure in computational materials science, the Novel Materials Discovery (NOMAD) Laboratory, which was co-founded by Claudia Draxl and has been online since 2014. The main challenge for FAIRmat is the integration of many different experimental characterization techniques and methods of material synthesis.
FAIR data enabling new horizons for materials research
M. Scheffler, M. Aeschlimann, M. Albrecht, T. Bereau, H.-J. Bungartz, C. Felser, M. Greiner, A. Groß, C.T. Koch, K. Kremer, W.E. Nagel, M. Scheidgen, C. Wöll, and C. Draxl
Nature 604 (2022) 635
DOI: 10.1038/s41586-022-04501-x
Contact:
Prof. Dr. Claudia Draxl
Institut für Physik / IRIS Adlershof
Humboldt-Universität zu Berlin
Tel.: 030 2093-66363
E-Mail: claudia.draxlphysik.hu-berlin.de
www.fairmat-nfdi.eu
Bremsstrahlung of black holes and neutron stars from quantum field theory
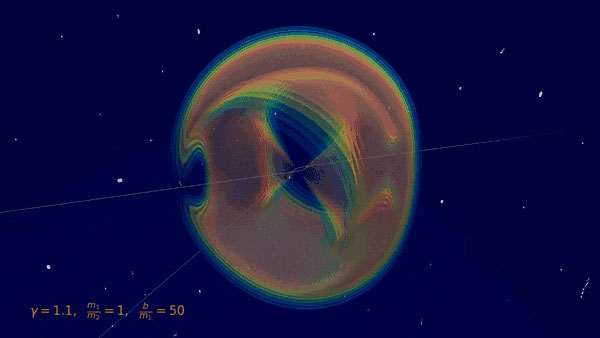
Fig 1: Visualization of the gravitational Bremsstrahlung from the scattering of two black holes (BSc thesis O. Babayemi)
When two massive objects (black holes, neutron stars, or stars) fly past each other, the gravitational interactions not only deflect their orbits, but also produce gravitational radiation, or gravitational bremsstrahlung, analogous to electromagnetism. The resulting gravitational waves of such a scattering event were calculated at leading order in Newton's gravitational constant in the 1970s using traditional methods of general relativity in an extensive series of four papers. Bremsstrahlung events are still out of reach for the current generation of gravitational-wave detectors because the signal is non-periodic and typically less intense. Nevertheless, they are interesting targets for future searches with future terrestrial and space-based observatories.
In the Quantum Field Theory lab around IRIS Adlerhof-member Prof. Plefka, a new approach to determining these waveforms (Fig. 1) and the deflections using methods of perturbative quantum field theory was recently developed, which proves to be significantly more efficient than the traditional approaches. It is based on a hybrid quantum field theory, in which the black holes (or stars) are idealized as point particles and interact with the gravitational field. The calculation is then based on a systematic diagrammatic expansion using Feynman graphs. I.e. the methods that were originally developed for the scattering of elementary particles can now also be used in astrophysical scenarios.
With this innovative method - the "Worldline Quantum Field Theory“ approach - our understanding of this fundamental physical process was recently significantly extended resulting in a series of three publications in the Physical Review Letters. In [1], the results from the 1970s were reproduced in a far more efficient way; this only required the calculation of three Feynman graphs (Fig. 2). In [2] the waveform could be extended to the case of rotating black holes and neutron stars. In a recent publication [3], the scattering angles and deflections in momenta and rotations due to the scattering process at the next-next-leading order of the gravitational constant were determined for the first time. Elaborated techniques for calculating Feynman integrals were used here. Interestingly, the rotational degrees of freedom of the black holes are described in this new formulation with a supersymmetric world line theory [4], which was originally developed in extensions of the Standard Model of particle physics.
This research was performed in the context of the DFG Research Training Group 2575 "Rethinking Quantum Field Theory“, where innovations in quantum field theory are developed in cooperation with the Max-Planck Institute for Gravitational Physics and the Helmholtz-Centre DESY.
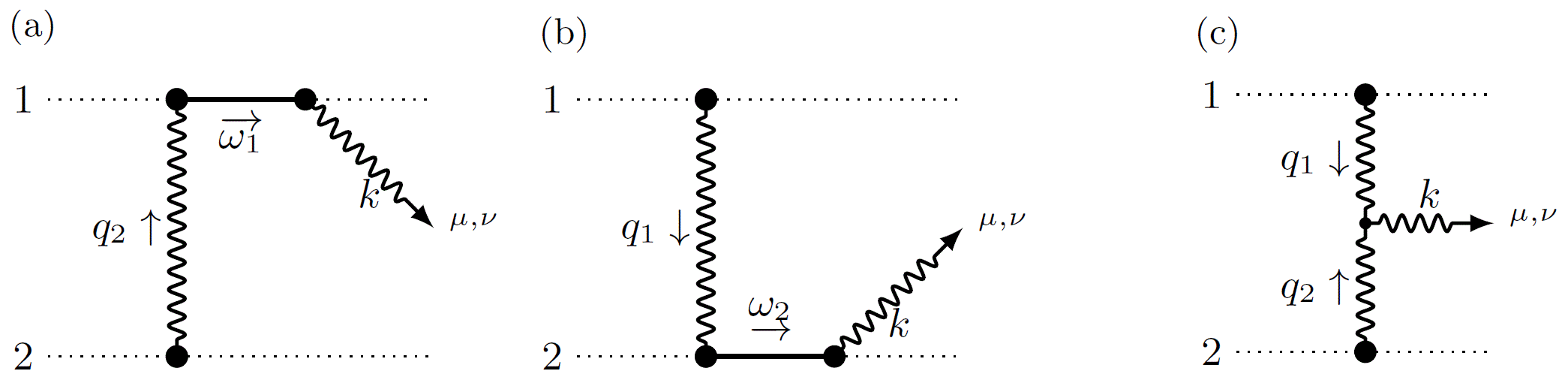
Fig 2: Feynman graphs to determine the waveform. The dotted lines represent the black holes, the waves represent gravitational radiation, and the lines represent fluctuations in the black hole's orbit.
Publikations:
| [1] |
Classical Gravitational Bremsstrahlung from a Worldline Quantum Field Theory
G. U. Jakobsen, G. Mogull, J. Plefka, and J. Steinhoff
Phys. Rev. Lett. 126 (2021) 201103
arxiv: 2101.12688 OPENACCESS |
|
[3] |
Conservative and radiative dynamics of spinning bodies at third post-Minkowskian order using worldline quantum field theory
G. U. Jakobsen and G. Mogull
erscheint in PRL
arxiv: 2201.07778 OPENACCESS
|
| [2] |
Gravitational Bremsstrahlung and Hidden Supersymmetry of Spinning Bodies
G. U. Jakobsen, G. Mogull, J. Plefka, and J. Steinhoff
Phys. Rev. Lett. 128 (2022) 011101
arxiv: 2106.10256 OPENACCESS |
|
[4] |
SUSY in the sky with gravitons
G. U. Jakobsen, G. Mogull, J. Plefka, and J. Steinhoff
JHEP 2201 (2022) 027
arxiv: 2109.04465 OPENACCESS |
Further Informationen:
Videos on Youtube (from BSc thesis of O. Babayemi)
MORE SCIENTIFIC HIGHLIGTHS
|